



Abstract Immobilized enzymes have become the subject of considerable interest due to their excellent functional properties such as reusability, cost-effectiveness, and optimality during the past decades. Enzyme immobilization technology is not only used in industrial processes, but also a component technology of products for medical diagnostics, therapy, food industry, bio energy, and biomaterial detection. In this review, new methods for enzyme immobilization are introduced, and the advantages and disadvantages of a variety of techniques in enzyme immobilization will be also discussed.
Keywords: Enzyme, Immobilization, Immobilized enzyme, Enzyme immobilization methods
I. Introduction
Most enzymes are relatively unstable, and have high production and separation costs, displaying a disadvantage in that the recovery of active enzymes in the reaction mixture after use is technically very difficult [1]. Immobilized enzymes have received great attention from those who wish to use the enzyme immobilization technology for specific purposes in the medical and industrial sectors [2]. The term “Immobilized enzymes” is defined as “Enzymes that is physically attached to specific solid supports and thus confined, and which can be used repeatedly and continuously while maintaining their catalytic activities” [3]. In recent years, enzymatic productivity has been rapidly growing through the improvement of genetic engineering technology, microbial cultivation technology and wild type strain screening technology in parallel with the understanding of enzymatic biosynthesis mechanisms [2,4-6]. The use of immobilized enzyme in biotechnology has some advantages such as [7,8]: First, a single batch of enzymes could be used multiply or repetitively. Second, immobilized enzymes are usually more stable than mobile enzymes. Third, the reaction could be controlled rapidly by removing the enzyme from the reaction solution. An additional advantage is the easy separation of the enzyme from the product so that contamination could be avoided. Also, the use of immobilized enzyme allows the development of a multi-enzyme reaction system. Over the past decades, biochemical and biophysical studies have been actively performed for the purpose of enhancing the stability and activity of enzymes hrough immobilization of enzymes [9]. The introduction of immobilized enzyme catalysts has greatly improved the technical performance of industrial processes, thereby increasing productivity and economic efficiency [10]. In this review, recent advances and novel strategies in methods of enzyme immobilization are briefly discussed, thereby providing a helpful information for choosing the appropriate immobilization scheme to improve the stability and activity of an enzyme of interest.
II. Techniques of Enzyme Immobilization
In an enzymatic reaction, an enzyme acts as a biological catalyst that promotes the reaction rate and is not worn out during the reactions. Thus, it allows for the repeatedly use of enzyme as long as the enzyme remain active. So far, a variety of immobilization procedures have been developed for immobilizing enzyme on a solid surface. The different enzyme immobilization methods are grouped as follows and listed in Figure 1 below. 1. Adsorption; 2. Covalent bonding; 3. Entrapment; 4. Cross-linking.
1. Adsorption
Enzyme immobilization onto biosensor transducer solid surface by adsorption is one of the most straightforward methods in immobilization. The adsorption mechanisms 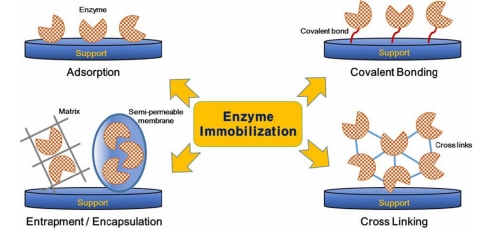
Figure 1. Various methods of enzyme immobilization.
are based on weak bonds such as Van der Waal’s forces, electrostatic and hydrophobic interactions [11]. Enzyme is dissolved in solution and the solid support is placed in contact with the enzyme solution for a fixed period of time under suitable conditions which sustain enzyme activity. The unadsorbed enzyme molecules are then removed from the surface by washing with buffer. Immobilization by adsorption is a simple and economical process which is reagent-free, low cost and is generally non-destructive toward enzyme activity because it does not involve any functionalization of the support. Nevertheless, this technique presents drawbacks: enzymes are loosely bound to the support by weak physical bonding so that changes in temperature, pH or ionic strength may result in enzyme desorption/leaching [12]. In addition, biosensors based on adsorbed enzyme suffer from poor operational and storage stability because apart from enzyme leaching, non-specific adsorption of other proteins or substances on the transducer surface may cause contamination and interference to signal. Immobilization by adsorption is commonly divided into 3 sub categories as follows: 1. Physical adsorption; 2. Electrostatic binding; 3. Hydrophobic adsorption.
1.1 Physical adsorption
This immobilization strategy has widely been used to develop enzymatic biosensors. Physical adsorption requires soaking of the support into a solution of the enzyme and incubating for a certain time period (hours) to allow physical adsorption to occur. Enzymes are absorbed to the supporting matrix through weak non-specific forces such as hydrogen bonding, Van der Waals forces, or hydrophobic interactions. However, these relatively weak nonspecific forces may result in a reversible process where enzyme leakage from the matrix could occur by changing the conditions that influence the interaction strength (e.g., pH, ionic strength, temperature, or polarity of the solvent). In another approach, enzyme solution is allowed to dry on the electrode surfaces followed by rinsing enzymes that are not adsorbed away [13,14]. In general, the method offers simplicity, surface regeneration and cost-saving capability however is time and reagent consuming. In addition, the obtained immobilized enzyme layer does not have homogeneity in bound molecule orientation hence substrate binding to enzyme active sites maybe hindered.
1.2 Electrostatic binding
To utilize electrostatic force in enzyme immobilization, the reaction solution pH and the isoelectric point of enzyme are the two parameters of consideration. The surface of enzyme molecules may bear positive or negative charge depending on the comparative difference between the isoelectric point of the enzyme and the pH value of the solution so that enzyme could be immobilized onto the
opposite charged surface via ionic and strongly polar interactions. Two common electrostatic adsorption immobilization techniques are layer-by-layer deposition and electrochemical doping, which have been widely employed to develop enzymatic biosensors.
1.2.1 Layer-by-layer deposition
Layer by layer (LBL) deposition used in enzyme immobilization is a thin-film fabrication method in which opposite charge layers of enzyme and materials are alternatively produced on top of each other on a solid support with wash steps in between. The deposition
process is simply done by dipping a cationic/anionic charged substrate into an aqueous solution of anionic/ cationic polyelectrolyte alternatively. The coated substrate is then rinsed and dipped into a solution of cationic/anionic enzyme. These alternating deposition processes are carried out repeatedly until a desired number of layers is obtained. Multilayers of opposite-charged layers is formed relying on electrostatic interactions, hydrogen bonding, coordination bonding, charge transfer, molecular recognition, hydrophobic interactions, or a combination of these. LBL deposition has widely been used for its simplicity and high biocompatibility. Utilize inherent charge property of enzyme molecule surface, the method offers an easily-controllable approach for enzyme immobilization, where layer thickness and layer structure can be easily modified. In LBL assembly technique, fabrication could be done at mild conditions with a small amount of material thus it is a cost-effective preparation method. In addition, the obtained multilayer thin film have special uniformity and stability thus help minimizing enzyme denaturation. The only disadvantage is however, overcharging of surface, substrate or product may
cause kinetics distortion due to partitioning or diffusion phenomena, and consequently change the pH stability of enzyme. Basically, the directions for the application of LBL assembly technique may vary depending on the type of material which deposits on the enzyme layer/film such as : the use of polyelectrolytes (conductive polymers) [15] and particles [16], biomacromolecules [17-19], dyes [20], dendrimers [21]. Among the others, polyelectrolyte has gained more preference in enzyme immobilization. Polyelectrolytes are water soluble polymers which carry ionic charge along the polymer chain. A polyelectrolyte could be either cationic (polycations) or anionic
(polyanions). Polycations that have mainly been used in LBL films include poly(allylamine) (PAA), poly(l-lysine) (PLL), poly(ethyleneimine) (PEI), poly(dimethyldiallylammonium chloride) (PDDA), poly(allylamine hydrocholoride) (PAH) and chitosan (CHIT). The most commonly used polyanions are poly(-stryrenesulfonate) (PSS), poly(vinylsulfonate) (PVS), poly(anilinepropanesulfonic acid) (PAPSA), poly(acrylic acid)(PAA) and poly(methacylic acid) (PMA) [22].
1.2.2 Electrochemical doping
Enzymes can also be immobilized into the conductive polymer film by electrochemical doping which occurs during the oxidation or reduction process of the polymer. During oxidation or reduction process, the polymer becomes positively/negatively charged thus charged
enzymes respectively could be incorporated into that conductive polymer. For example, a biosensor based on electrochemical doping immobilization of galactose oxidase was developed for galactose monitoring [23]. The reduced polyaniline film was immersed in galactose
oxidase solution and then oxidized at 0.60 V. Galactose oxidase was immobilized in the polyaniline film during the oxidation process: since the fibril diameter of polyaniline is about 2000 A while enzyme particle diameters are in the range 100-1000 A [24]. The biosensor showed a linear detection range with galactose concentration between 0.2 and 6 mmol dm−3
. A similar approach was applied for human blood glucose determination by immobilizing GOD on polyaniline film by doping [25]. GOD bearing a negative charge is doped into the polyaniline film during the oxidation process. The fabricated glucose biosensor has
a high operational stability and long storage stability (36 months)
1.3. Hydrophobic adsorption
Another immobilization approach is the use of hydrophobic interactions between the support and enzyme molecules. The interaction formed by the displacements of water molecules from support surface material and enzyme molecules surface during immobilization as a result of entropy gain [26]. The interaction strength heavily depends on the hydrophobicity of both the adsorbant and enzyme.
Experimental variations such as pH, temperature and concentration of salt during enzyme immobilization or the size of hydrophobic ligand molecule and the degree of the support substitution could be adjusted to regulate the hydrophobic interactions between the enzyme and support [27,28]. Successful example of reversible immobilization by hydrophobic adsorption onto hexyl-agarose carriers has been reported for β-amylase and amyloglucosidase [29,30].
2.2. Covalent bonding
Enzyme immobilization by covalent binding is one of the most widely used methods, in which stable complexes between functional groups on enzyme molecules and a support matrix are formed through covalent bondings. The functional group present on enzyme, through which a covalent bond with support could be established, should be non-essential for enzymatic activity which usually involves binding via the side chains of lysine (ϵ-amino group), cysteine (thiol group) and aspartic and glutamic acids (carboxylic group). The enzyme functional groups that could be utilized in covalent coupling include: Amino group, carboxylic group, phenolic group, sulfhydryl group,
thiol group, imidazole group, indole group and hydroxyl group [31]. The binding procedure of enzyme to the solid support generally goes through two stages: (1) activation of the surface using linker molecules such as glutaraldehyde or carbodiimide and (2) enzyme covalent coupling to the activated support. Linker molecules are multifunctional reagents (glutaraldehyde or carbodiimide) act as the bridge
between surface and enzyme via covalent bonding. While the first group matches the immobilization surface and forms a so-called self-assembled monolayer (SAM), the second ground bound to preactivated support then forms a covalent bond with the enzyme. Different linkers are used for different surfaces (inorganic material, natural or synthetic polymer, membranes) and immobilization protocols (directly onto the transducer surface or onto a thin membrane fixed onto the transducer). Covalent immobilization provides strong bindings between enzymes and support matrix and therefore little leakage of enzyme from the support may occur. In
addition, high uniformity of the SAM layer and good control of the immobilized enzyme amount are the other advantages. In covalent attachment, there is a high risk of enzyme denaturization when most enzymes must go through chemical modifications to possess functional group. In addition, the method requires high volume of bioreagent but only small amounts of enzymes may be immobilized
(~0.02 grams per gram of matrix). The immobilization procedure largely increases enzyme stability but decreases enzyme activity in affinity reaction and is poorly reproducible [32]. In comparison to adsorption, covalent bonding requires longer incubation time, since the
formation of the SAM and the subsequent linkage of the enzymes to it take several hours. The process is also more complex and care has to be taken to ensure chemical purity so that the SAM is obtained in high homogeneity. The most used procedures to covalently immobilize enzyme on functionalized surface (through the activations of carboxylic group and amino group) are briefly described
below.
2.1. Activation of carboxylic groups
A carbodiimide is a functional group (formula RN=C=NR) which allows the binding between the carboxyl groups (-COOH) of a support and the amino function (-NH2) of an enzyme. In order to improve immobilization efficiency, N-hydroxysuccinimide (NHS) could be associated to carbodiimide prior to enzyme covalent coupling step.
2.2. Activation of amino groups
The binding between an amine functionalized support and carboxyl functionalized enzyme could also be done with carbodiimides. Alternatively, glutaraldehyde could be used as the activating agent for enzyme immobilization. Firstly, Schiff-base reaction occurs between amine functionalized support and an aldehyde group of glutaraldehyde and then, the second aldehyde group of glutaraldehyde covalently bind to an amine functionalized enzyme.
2.3. Chemisorption
The principle of this immobilization method based on a strong affinity and semi-covalent bond between thiol group
(-SH) and gold substrates (Au). Thus, thiol-containing enzymes, such as oxidoreductases and isomerases which contain double-catalytic site cysteine residues, could be immobilized on gold surface via the thiol groups of their amino acid residues. These thiol-containing enzymes are either in native forms or obtained through chemically modification or genetic engineering techniques, in order to
provide them with reactive thiol groups. A detailed paper on immobilization of enzyme via their thiol group could be found here [33]. Alternatively, thiol containing enzymes can be immobilized onto supports, which fixed with reactive disulfides or disulfide oxides, through a thiolcontaining bifunctional linker which, on one end, forms disulfide bonds (S–S) to the surface, and on the other end,
provides N-hydroxysuccinimide (NHS) groups that can react with the free amino groups on the enzyme.
3. Entrapment
In entrapment immobilization, enzyme is not directly attached to the support surface but entrapped within a polymeric network which allows only the traverse of substrate and products but retains the enzyme hence enzyme diffusion is constrained. Entrapment immobilization process is conducted through two steps: (1) mixing enzyme into a monomer solution, followed by (2) polymerization of
monomer solution by a chemical reaction or changing experimental conditions. As an enzyme is physically confined within a polymer lattice network, the enzyme does not chemically interact with the entrapping polymer. The method thus could improve enzyme stability and minimize enzyme leaching and denaturation. Another advantage of the method is the capability to optimize microenvironment for the enzyme by modifying the encapsulating material to have the optimal pH, polarity or amphilicity. However, a limitation of the method is the mass transfer resistance occurred as polymerization extension tends to increase the gel matrix thickness, substrate for this reason can not diffuse deep into the gel matrix to reach the enzyme active site. Furthermore, the entrapped enzymes are likely to suffer from leakage if the pores size of the support matrix is too large. The method also has low enzyme loading capacity and the support material could be corrupted as effects of polymerization. There is a variety of procedures used in entrapment immobilization depending on type of entrapment such as electropolymerization, photopolymerization, sol-gel process for lattice or fiber type and microencapsulation for
microcapsule type.
3.1. Electrochemical polymerization
Electrochemical polymerization (or electropolymerization) is a simple approach in which an appropriate potential or current is applied into a solution containing both enzyme and monomer molecules. The oxidization or reduction reactions of monomers occurred in the solution at electrode surface could then generate reactive radical species whichcouple together and finally form an adherent polymer at the electrode surface. Enzyme molecules that are present in the solution close by the electrode surface are trapped inside the growing polymer network as polymerization process propagates. The first step in the polymerization process is the oxidation of the monomer to generate a radical cation which then could either react with a neutral monomer or with another similar radical to form a dimer. The formed dimers then undergo further oxidation process and coupling reactions to generate oligomers and finally produce an insoluble polymer deposited on electrode surface. Most of electropolymerized films used for enzyme immobilization are electronically conducting polymers such as polyaniline, polypyrrole or polythiophene, pyrroles, thiophenes and polyindole. In addition, other materials such as redox conductors as in the case of metal poly(pyridine) complexes and non-conducting (insulating) polymer, as in the case of phenols, 1, 2-diaminobenzene are also applicable for electropolymerization. However, electropolymerized films of conducting polymers have been predominantly employed in various sensor types. The distinct advantages of conducting polymers over other materials is the
conductivity which helps control the deposition site and thickness of the polymer films easily because the continuing growth of the polymer thickness is exclusively done on electrode surface and driven electrochemically by the applied potential and propagation time. In comparison to manual deposition, electrochemical deposition by mean of polymerization has better controls over the homogeneity
and thickness of the polymer film because the homogeneity could be evaluated from the formation of a diffusion barrier over the film and the thickness could be measured by the charge transferred during film formation. Other parameters that could affect the nature and morphology of the polymer film are the choice of solvent, counter-ion, and conditions used in the electrochemical polymerization such as
temperature, monomer concentration and the electrolyte chain length of the polymer. Electrochemical polymerization offers a simple one-step method which could produce homogeneous films by an easy control of applied potential. On the electrode, high enzyme activity is retained because there is no interaction between enzyme and monomer during the polymerization process which propagates
exclusively on the electrode surface. However, there are some criteria that one should follow for a reproducible immobilization of enzyme as follows: The polymerization process should be carried out in an oxygen-free environment with proper polymerization solution. The
change of pH and charge of the polymer due to protons liberation during the polymerization reaction may affect enzyme activity and should be compensated by incorporation of anions from the electrolyte. The drawbacks of this method is the requirement of high concentrations of monomer (0.05-0.5 M), enzyme (0.2-3.5 mg ml−1) and film deterioration resulted from overoxidation process which
happens due to monomer depletion in electrode surrounding solution.
3.2. Photopolymerization
In photopolymerization process-based enzyme immobilization the use of liquid, photopolymers (radiation curable resins) and enzyme solution are required. The photopolymerization reactions are chain-growth polymerizations which are initiated when the photopolymers exposed to light in the ultraviolet or visible region of the electromagnetic spectrum. Upon light exposure, these photopolymers undergo chemical reactions for cross-linking of molecules resulting in the hardening of the material. The reactant monomer may absorb light either directly or through an energy transfer from a photosensitizer. In general a photopolymerization process goes through 4 stages of nitiation, propagation, termination and chain transfer steps. Also, the polymerization process has been used to entrap enzymes for poly(vinyl alcohol)-bearing styrylpyridinium groups (PVASbQ), a soluble pre-polymer bearing photocrosslinkable groups, which has largely been used to entrap enzymes since its first synthesis [34-36]. In the propagation of photopolymerization reactions, light irradiation is required to triggers additional cross-linking reactions between comonomers to form oligomers and finally generate an insoluble polymer
3.3. Sol-gel process
The sol-gel process is based on the ability to form metaloxide, silica, and organosiloxane matrices of defined porosity by the reaction of organic precursors at room temperature [37]. There are two generic methods of the sol-gel technique depending on the types of starting materials (precursors) used: colloidal method, and polymeric (or alkoxide) route. In enzyme immobilization the latter method is commonly employed. The route involves 2 following steps: (1) Suspending or dissolving the metal alkoxide precursor(s) such as tetramethoxysilane or methyltrimethoxysilane in a suitable liquid (acidic pH in the presence of water) for hydrolyzation to produce silanol (Si-OH) groups. (2) The hydrolized precursor is then activated by the addition of a base (such as potassium hydroxide) to initiate condensation reactions between silanol moieties resulting in the formation of siloxane (SiO-Si) polymers. As the network grows and ages with time and temperature, the viscosity of the liquid increases at an exponential rate until gelation occurs. As a result, a matrix is created in which the enzyme molecules are enclosed within the network [38-41]. Sol-gel formation is a popular immobilization method that results in a stable nanoporous material where enzyme activity is preserved and biosensor sensitivity is enhanced owing to high encapsulation concentration and mild immobilization conditions. However the method may suffer from extra cost of precursors and matrix inhomogeneity due to fracture during gelation drying and precipitation of oxides during sol formation.
3.4. Micro-encapsulation
Immobilization by encapsulation represents an entrapment method in which enzymes are enclosed in a spherical semipermeable membrane. The membrane may be polymeric, lipoidal, lipoprotein based or non-ionic in nature. In general, there are two methods for microencapsulation: (1) Coacervation (or phase separation) in which enzyme microdroplets are separate out in a water immiscible solvent and (2) Interfacial polymerization in which a monomer is made to be polymerized at the interface of two immiscible substances (a hydrophobic monomer and another monomer which is dispersed in a water immiscible solvent). This polymerization process results in the agents through surface complementarity which helps increase stability [43]. However, the use of glutaraldehyde could result in severe enzyme modifications and possibly lead to enzyme conformational changes and loss of activity. For this reason, inert proteins like gelatin, bovine serum albumin (BSA) may be added during the immobilization process to minimize this drastic modification of enzymes [44].
III. Conclusions
Advantages that an efficient enzyme immobilization could bring about are the repetitive use of a single batch of enzymes, improved stability, abilities to stop the reaction rapidly by the removal of enzyme from the reaction solution, easy separation of the enzyme from the product and the avoidance of enzyme-product contamination. In addition, multi-enzyme reaction systems for multiple analyte detection could be developed for biosensor applications based on the enzyme immobilization. The choice of immobilization method in biosensors depends on many factors, such as the nature of the biological element, the transducer type, the physicochemical properties of the analyte and the biosensor operating conditions.